





Abstract
The transcription factor CsgD plays a key role in the control of biofilm formation in Escherichia coli by controlling the production of curli fimbriae and other biofilm components. In concert with its regulatory role, the promoter for the csgD operon is under the control of more than 10 regulatory factors, each monitoring a different condition or factor in stressful environments. Previously, we classified three factors (OmpR, RstA and IHF) as activators and two factors (CpxR and H-NS) as repressors, and found novel modes of their interplay. Here we describe an as yet uncharacterized regulator, MlrA, that has been suggested to participate in control of curli formation. Based on both in vivo and in vitro analyses, we identified MlrA as a positive factor of the csgD promoter by directly binding to its upstream region (−113 to −146) with a palindromic sequence of AAAATTGTACA(12N)TGTACAATTTT between the binding sites of two activators, IHF and OmpR. The possible interplay between three activators was analysed in detail.
Introduction
Under stressful conditions in nature, planktonic single-cell Escherichia coli transforms into multicellular biofilm through adhesion to solid surfaces and cell–cell interactions using extracellular matrix compounds such as cellulose and curli fimbriae (Prigent-Combaret et al., 2000; Chapman et al., 2002; Beloin et al., 2008; Gualdi et al., 2008; Wood, 2009). The synthesis of csgBA-encoded curli is induced at low temperatures and under low osmolarity during stationary phase growth (Barnhart & Chapman, 2006). Expression of csgBA is under the control of a positive regulator, CsgD, which is also involved in the regulation of cellulose production and peptidase synthesis (Prigent-Combaret et al., 2001; Brombacher et al., 2003, 2006; Chirwa & Herrington, 2003; Gerstel & Romling, 2003). In concert with the regulatory role of CsgD as the master regulator of biofilm formation under stressful conditions, the major sigma RpoD and stationary phase-specific RpoS participate in transcription initiation from two promoters of the csgD operon (Robbe-Saule et al., 2006; Gualdi et al., 2007; Ogasawara et al., 2007a, 2010). Furthermore, a number of transcription factors are involved in the regulation of the csgD promoter, including CpxR (Jubelin et al., 2005), Crl (Bougdour et al., 2004), H-NS (Gerstel et al., 2003), IHF (Gerstel et al., 2003, 2006), OmpR (Vidal et al., 1998; Gerstel et al., 2003, 2006), RstA (Ogasawara et al., 2007a), MlrA (Brown et al., 2001), RcsB (Ferrieres & Clarke, 2003; Vianney et al., 2005) and CRP (Zheng et al., 2004). On the basis of these observations, the csgD promoter is now recognized as one of the most complex promoters in E. coli (Ishihama, 2010).
As an initial step toward understanding the regulatory mechanisms of the csgD promoter by a number of transcription factors, we have classified some of these transcription factors into positive and negative regulators (Ogasawara et al., 2010). In addition, we and others have analysed the pair-wise interplay between RpoS and Crl (Robbe-Saule et al., 2006), IHF and H-NS (Gerstel et al., 2003; Ogasawara et al., 2010), OmpR and IHF (Gerstel et al., 2003, 2006; Ogasawara et al., 2010), CpxR and OmpR (Jubelin et al., 2005), CpxR and H-NS (Ogasawara et al., 2010), and RstA and IHF (Ogasawara et al., 2010). As an extension, here we focused on the regulatory role of an as yet uncharacterized transcription factor, MlrA (MerR-like regulator; renamed from YehV), which was identified as a novel positive regulator for the synthesis of curli fimbriae and extracellular matrix in E. coli and Salmonella typhimurium (Brown et al., 2001), although the mode of its action remains largely unidentified. For transcription initiation of the mlrA gene, not only is RpoD involved but so too is RpoS, and thus expression of the mlrA gene is induced in the stationary phase (Brown et al., 2001). MlrA contains a conserved N-terminal DNA-binding domain present in members of the MerR family, implying that MlrA is a DNA-binding regulatory protein. However, its C-terminal domain exhibits no similarity to any of the hitherto characterized MerR family members.
In the present study, we found that MlrA is indeed a DNA-binding transcription factor, and is involved in activation of the csgD promoter by binding between the promoter-distal IHF-binding site in hot-spot I and the OmpR-binding site in hot-spot II for interaction with transcription factors. Interplay between three activators, MlrA, OmpR and IHF, was also analysed with respect to control of this complex promoter.
Materials and methods
Bacterial strains
Escherichia coli K-12 wild-type K-12 BW25113 and mutant strains lacking the genes for transcription factors were the products of the Keio Collection, and were obtained from the E. coli stock centre of the National Institute of Genetics (see Supporting Information, Table S1). Escherichia coli BWcsgD and BWmlrA were KmS derivatives of JW1023 and JW2115, respectively, that were constructed using pCP20. Escherichia coli BL21(DE3) F-ompT hsd (rB− mB−) dcm gal λ(DE3) was used for expression and purification of the transcription factors used in this study. Escherichia coli MC4100 was used for construction of the csgD–lacZ, csgB-lacZ and mlrA-lacZ reporter vectors.
Expression and purification of transcription factors
For construction of plasmids (pMlrA and pOmpR) for expression of His-tagged MlrA and OmpR, DNA fragments corresponding to the coding sequences of their respective transcription factors were amplified by PCR using E. coli W3110 genome DNA as a template and pairs of primers, which were designed so as to hybridize upstream or downstream of each coding sequence (Table S2). After digestion with NdeI and NotI (note that the restriction enzyme sites were included within the primer sequences), PCR products were cloned into pET21a(+) (Novagen) between NdeI and NotI sites. The plasmid constructs were confirmed by DNA sequencing.
For protein expression, the plasmids were transformed into E. coli BL21 (DE3), and the transcription factors were overexpressed and purified as His-tagged forms (Igarashi & Ishihama, 1991; Yamamoto et al., 2005). In brief, E. coli BL21 (DE3) transformants were grown in 200 mL Luria–Bertani (LB) medium and at a cell density of 0.6 at 600 nm, and isopropyl-β-d-thiogalactopyranoside was added at a final concentration of 1 mM. After 3 h of incubation, cells were harvested by centrifugation, washed with lysis buffer (50 mM Tris-HCl, pH 8.0 at 4 °C, and 100 mM NaCl), and then stored at −80 °C until use. For protein purification, frozen cells were suspended in 3 mL lysis buffer containing 100 mM phenylmethylsulfonyl fluoride. Cells were treated with lysozyme and then subjected to sonication for cell disruption. After centrifugation at 20 400 g for 20 min at 4 °C, the resulting supernatant was mixed with 2 mL of 50% Ni-nitrilotriacetic acid agarose solution (Qiagen) and loaded onto a column. After washing with 10 mL of the lysis buffer, the column was washed with 10 mL of washing buffer (50 mM Tris-HCl, pH 8.0 at 4 °C, and 100 mM NaCl), and then 10 mL of washing buffer containing 10 mM imidazole. Proteins were eluted with 2 mL of an elution buffer (lysis buffer plus 200 mM imidazole), and peak fractions of transcription factors were pooled and dialysed against a storage buffer (50 mM Tris-HCl, pH 7.6 at 4 °C, 200 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol and 50% glycerol), and stored at −80 °C until use. Protein purity was checked by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE).
RpoD sigma and IHF were purified in native form without His-tag as described previously (Murakami et al., 1996; Azam & Ishihama, 1999).
Gel mobility shift assay
The gel shift assay was performed as described previously (Ogasawara et al., 2007a, b). In brief, probes were generated by PCR amplification of the csgD promoter region using a pair of primers (Table S2; csgB-S1F primer and 5′-FITC-labelled csgB-FITC-R primer for CD7, and csgD-F and 5′-FITC-labelled csgD-FITC-R for CD6), and pRScsgD containing the recognition sequences by each transcription factor as a template, and Ex Taq DNA polymerase (Takara).
PCR products with fluorescein isothiocyanate (FITC) at their termini were purified by PAGE. For gel shift assays, mixtures of the FITC-labelled probes and purified transcription factors were incubated at 37 °C for 30 min in 12 μL of gel shift buffer (10 mM Tris-HCl, pH 7.8 at 4 °C, 150 mM NaCl and 3 mM magnesium acetate). After addition of a DNA dye solution, the mixture was directly subjected to 6% PAGE. Fluorescently labelled DNA in gels was detected using LAS4000 (Fuji Film).
DNase-I footprinting assay
Labelling of probe DNA with FITC was performed as described previously (Ogasawara et al., 2007a, b). Each 0.5 pmol of FITC-labelled probe was incubated at 37 °C for 30 min with various amounts of MlrA in 25 μL of DNase-I footprinting solution [10 mM Tris-HCl (pH 7.8), 150 mM NaCl, 3 mM magnesium acetate, 5 mM CaCl2 and 25 μg mL−1 bovine serum albumin]. After incubation for 30 min, DNA digestion was initiated by the addition of 5 ng of DNase I (Takara). The reaction was terminated by the addition of 25 μL of phenol. Phenol-treated products were precipitated with ethanol, dissolved in formamide-dye solution and analysed by electrophoresis on a 6% polyacrylamide gel containing 7 M urea using a Shimadzu slab gel electrophoresis system (DSQ-2000L; Shimadzu, Kyoto).
Primer extension assay
For preparation of total RNA for primer extension assays, overnight cultures were diluted 100-fold in 30 mL of YESCA medium (per litre: 1 g yeast extract, 10 g Casamino acids) (Pratt & Silhavy, 1998) and MlrA-overproducing E. coli cells were incubated for 5 h at 37 °C until the exponential phase (OD600 nm=0.5–0.6). RNA purification was carried out as described previously (Ogasawara et al., 2007a, b).
Primer extension analysis was performed using fluorescently labelled probes according to the protocol of Yamada (1998). In brief, 20 μg of total RNA and 1 pmol of 5′-FITC-labelled primer (csgD-FITC-R: 5′-GCACTGCTGTGTGTAGTAAT-3′) were mixed in 20 μL of 10 mM Tris-HCl (pH 8.3 at 37 °C), 50 mM KCl, 5 mM MgCl2, 1 mM each of dATP, dTTP, dGTP and dCTP, and 20 U of RNase inhibitor. The primer extension reaction was initiated by the addition of 5 U of avian myeloblastosis virus reverse transcriptase. After incubation for 1 h at 50 °C, DNA was extracted with phenol, precipitated with ethanol and subjected to electrophoresis on a 6% polyacrylamide sequencing gel containing 8 M urea. After electrophoresis, gels were dried and subjected to autoradiography using DSQ-500L (Shimadzu).
LacZ reporter assay of promoter activity
A 438-bp fragment of the csgD promoter, a 489-bp fragment of the csgB promoter and a 355-bp fragment of the dppB promoter upstream from the respective translation start site were prepared by PCR using E. coli K-12 KP7600 genome as a template and a pair of primers, csgD-EcoRI-F and csgD-BamHI-R2 for csgD, csgB-EcoRI-F and csgB-BamHI-R for csgB, and dppB-EcoRI-F and dppB-BamH1-R for dppB (Table S2). After digestion with EcoRI and BamHI, each fragment was ligated into pRS551 (Simons et al., 1987) at the corresponding sites. The resulting plasmids were transformed into E. coli MC4100 and the transformants were used as hosts for preparation of λRS45. Recombinant λ phages containing csgD –lacZ, csgB–lacZ or dppB–lacZ fusions were infected onto the wild-type, csgD or mlrA mutant E. coli strains (Table S1). Cells were cultured in LB medium or YESCA medium at 37 °C. When necessary, 100 μg mL−1 ampicillin and 50 μg mL−1 kanamycin were added. Escherichia coli transformants were grown in LB medium and subjected to a β-galactosidase assay with o-nitrophenyl-d-galactopyranoside as a substrate (Miller, 1972).
For monitoring the influence of MlrA on expression of the lacZ reporter plasmids, an arabinose-inducible MlrA-expression plasmid was constructed using pBAD18 vector (Guzman et al., 1995). In brief, a DNA fragment containing the MlrA-coding frame was prepared by PCR using E. coli KP7600 genome DNA as a template and a set of primer pairs (Table S2). After digestion with EcoRI and XbaI (note that the restriction enzyme sites were included within the primer sequences), the PCR-amplified fragments were inserted into pBAD18 at the corresponding sites to generate the MlrA-expression plasmid pBADmlrA. The plasmid construct was confirmed by DNA sequencing.
Congo red (CR) plate assay of curli formation
A CR plate assay was performed as described previously (Barnhart & Chapman, 2006). To determine curli assembly, each E. coli test strain was grown at 28 °C for 48 h on YESCA plates containing 50 μg mL−1 CR and 10 μg mL−1 Coomassie blue.
Results
Role of MlrA in control of the csgD promoter: LacZ reporter assay
As an initial attempt to identify the regulatory role of MlrA on the csgD promoter, we examined the possible influence of its overexpression on the csgD promoter–lacZ reporter fusion. In wild-type background, overexpression of MlrA led to an approximately 2.5-fold increase in csgD–lacZ expression (Fig. 1a). In the csgD knockout mutant, this increase was more than 4.5-fold (Fig. 1b), indicating that MlrA is a positive factor of the csgD promoter. As overexpression of MlrA did not affect the promoter of the divergently transcribed csgBC operon, the effect of MlrA on curli production is solely attributable to activation of the csgD promoter, ultimately leading to activation of the csgB promoter. The expression level of MlrA in both wild-type E. coli and the csgD mutant was essentially the same as detected by immunoblot analysis of whole-cell lysates (data not shown).
![Effect of MlrA on the csgD, csgB and dppB promoters. (a) Escherichia coli BWWcsgD [wild-type BW25113(λcsgD–lacZ)], (b) BWcsgDcsgD [mutant BWcsgD(λcsgD–lacZ)], (c) BWcsgDcsgB [mutant BWcsgD(λcsgB–lacZ)] and (d) BWmlrAdppB [mutant BWmlrA(λdppB–lacZ)] strains were transformed with pBAD18 (lane 1) or pBADmlrA (lane 2), and grown on YESCA medium in the presence of 0.02%l-arabinose. In mid-exponential phase growth (OD600 nm=0.4–0.5), aliquots were subjected to a β-galactosidase assay.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsle/312/2/10.1111_j.1574-6968.2010.02112.x/3/m_FML_2112_f1.jpeg?Expires=1716408222&Signature=lwWKWNN7CjaNyuDB1aoGfz2XhJkP5lWHkYvVD7fuacgs0ZrUvbCwpjSOMHi2gLPlbBPgu5PwOo7SMhl2SkpM41NqTA4wSVU1B~6LTQ6tCuJ4gPYpZpEOYl4mHTYcOmBk2TZrnqRK8iTYk7Bbmj-JdW-3RQsvKRmBdvtFqGba43rdys6od-naBafx2wkNuUAZRWW63hv958OaDTN2fz5NnQeoYBoACW0hhnRBA83XNFKhe0D6JOrfoF~vjFc5MM~JeAVwSRgp8JsYz~POBg1Vn67DPVMhfy-C62SsM1h-pbj1sTXVdOF7wa9k-T~OGOv1EjHXzeTQcl9P1djeKUROAQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of MlrA on the csgD, csgB and dppB promoters. (a) Escherichia coli BWWcsgD [wild-type BW25113(λcsgD–lacZ)], (b) BWcsgDcsgD [mutant BWcsgD(λcsgD–lacZ)], (c) BWcsgDcsgB [mutant BWcsgD(λcsgB–lacZ)] and (d) BWmlrAdppB [mutant BWmlrA(λdppB–lacZ)] strains were transformed with pBAD18 (lane 1) or pBADmlrA (lane 2), and grown on YESCA medium in the presence of 0.02%l-arabinose. In mid-exponential phase growth (OD600 nm=0.4–0.5), aliquots were subjected to a β-galactosidase assay.
Identification of the MlrA-binding site on the csgD promoter
To gain insight into how the csgD promoter is activated by MlrA, we next examined the DNA-binding activity of purified MlrA and determined the site of recognition and binding on the csgD promoter. Gel-shift assays using various types of csgD probes covering various portions of the csgD promoter and its upstream region (for DNA segments see Ogasawara et al., 2010), only the CD6 (−67 to −335 from csgDp1) probe was found to bind stable complexes with MlrA (Fig. 2a), but MlrA did not bind to CD7 (−335 to −615) (Fig. 2b).
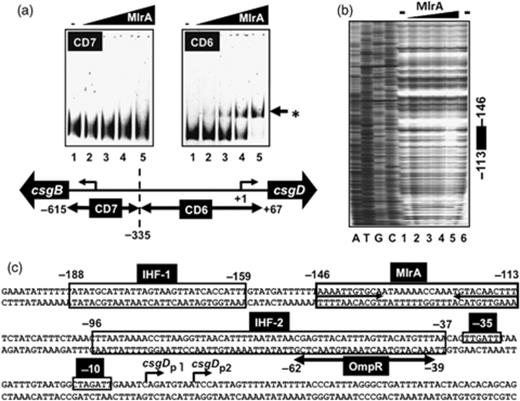
Gel-shift and DNase I footprinting analyses of the csgD promoter. (a) Fluorescently labelled DNA probes (CD6 and CD7, 0.5 pmol each) were incubated at 37°C for 30 min with purified MlrA (lanes 1–5: 0, 1.25, 2.5, 5.0, and 10 pmol) and directly subjected to PAGE. Lower panel: the location of CD6 and CD7 probes along the csgD promoter. Numbers at the probe ends represent the distance from the csgD P1 transcription start site. (b) Fluorescently labelled DNA probe of the csgD promoter fragment (1 pmol) was incubated with increasing concentrations of purified MlrA (lanes 1–5: 0, 2.5, 5, 10 and 20 pmol; lane 6, none), and then subjected to a DNase I footprinting assay. The filled bar at right indicates the region protected by MlrA from DNase digestion. Numbers at the bar ends represent the distance from the csgD P1 transcription initiation site. (c) Transcription factor-binding sequences along the csgD promoter were determined by DNase-footprinting assay. Boxed sequences represent the binding sites of IHF (site-I) and MlrA while the OmpR-binding sequence is shown by a bar below the sequence. The inverted repeat sequence within the MlrA-binding site is shown by arrows. The −10 and −35 signals show the promoter P1 of the csgD promoter. Numbers at both ends of each line represent the distance from csgD P1 transcription start site.
For identification of the MlrA-binding sequence, DNase I-footprinting mapping was performed using the purified His-tagged MlrA protein and the csgD promoter DNA fragment. A single unique protection sequence was identified (Fig. 2b and c) covering a 33-bp sequence (−113 to−146) including an inverted repeat sequence of AAAGTTGTACA(12N)TGCACAATTTT (Fig. 2c). This MlrA-binding sequence consisting of a 11-bp-long inverted repeat with a 12-bp interval is unique with respect to the length (total 32 bp long) of the consensus sequence of other characterized transcription factors from E. coli (Ishihama, 2010). The MlrA-binding site is located between the promoter-distal IHF-binding site in the transcription factor-binding hot-spot I and the OmpR-binding site in the hot-spot II along the csgD promoter (Ogasawara et al., 2010). After searching the predicated MlrA-box sequence along the whole E. coli genome, we identified four additional MlrA-binding target sequences (Fig. 3a), which all carry a well-conserved MlrA-box sequence (Fig. 3b). A gel shift assay indicated MlrR binding to the predicted target sequences (data not shown). Using the promoter–lacZ reporter fusion system, increased LacZ activity was detected in the mlrA mutant only when MlrA was overexpressed (for instance, see the dppB promoter in Fig. 1d). We therefore concluded that the newly identified genes are under the direct control of MlrA. Note that besides the csgD promoter, the genes for two additional transcription factors, CadC and YrbA, are also under the control of MlrA. If this is the case, MlrA is located at a higher level in the hierarchy of the transcription factor network (Ishihama, 2010).
![Prediction of regulation targets of MlrA. (a) After searches for the inverted repeat sequence [AAAATTTGTACA(12N)TGTACAATTTT] of MlrA binding in the entire Escherichia coli genome, four additional sites were identified, which have two to five mismatches. For the genes shown in blue, MlrA box-like sequences are located at promoter-proximal upstream regions. (b) The MlrA box-like sequences on the predicted MlrA target genes are aligned.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsle/312/2/10.1111_j.1574-6968.2010.02112.x/3/m_FML_2112_f3.jpeg?Expires=1716408222&Signature=V1yNajzWtrGUpuFYABX8uif9iQ~3KHvu01GpHxj7uoek2x9XBF~eEzPE2YTP3WqJbLj5Letfig2fQUgK9hQsu3l6pfw~5PfgcGVaub22FNDH7yHrmrRceIx2jvfD1FPHZw1jFHZEmTvHl-umyO7II9COEc~9HFgS2aCX1PDHXai2PdcgjotIgZp0exWRmGfcVDZ-7nTklfQG6xOL2w~792GMCQ7K-zlEoA31PTpH4LwJjQd7jCz2pHO~Ib0Hfle5hzeBlkvOQOUDO2WwPzYbJrjSgINbsXcTdqsGWfRRW2U9AqKTA~Spv2pdFGngXcqpxwGZN3L6oIvX0wCRXN1VvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Prediction of regulation targets of MlrA. (a) After searches for the inverted repeat sequence [AAAATTTGTACA(12N)TGTACAATTTT] of MlrA binding in the entire Escherichia coli genome, four additional sites were identified, which have two to five mismatches. For the genes shown in blue, MlrA box-like sequences are located at promoter-proximal upstream regions. (b) The MlrA box-like sequences on the predicted MlrA target genes are aligned.
Role of MlrA in control of the csgD promoter: primer extension assay
To confirm the activating role of MlrA on the csgD promoter, we next measured csgD mRNA using the primer extension assay. The level of csgD P1 mRNA was low in cells at steady-state at the exponential phase of growth in LB medium (Fig. 4, lane 1). When MlrA was overexpressed using arabinose-inducible expression vector, csgD mRNA markedly increased even in the wild-type E. coli (Fig. 4, lanes 1 and 2). The enhancing effect of MlrA overexpression was also examined for a set of E. coli mutants, each lacking one of the transcription factors that are involved in the regulation of the csgD promoter (Ogasawara et al., 2010). In the presence of MlrA overexpression, an increase of csgD mRNA was observed in the mutants lacking Crl (Fig. 4, lanes 5 and 6), H-NS (Fig. 4, lanes 7 and 8), RstA (Fig. 4, lanes 11 and 12), CpxR (Fig. 4, lanes 13 and 14) and RcsB (Fig. 4, lanes 15 and 16), indicating that MlrA functions independent of these regulators. In contrast, however, enhancement of the csgD promoter was not detected for mutants lacking OmpR (Fig. 4, lanes 3 and 4) and IHF (Fig. 4, lanes 17–20), both binding near the MlrA-binding site (see Fig. 2; also see Fig. 6). One explanation for the lack of csgD promoter activation in the mutants lacking OmpR and IHF is that MlrA requires these two activators for function.
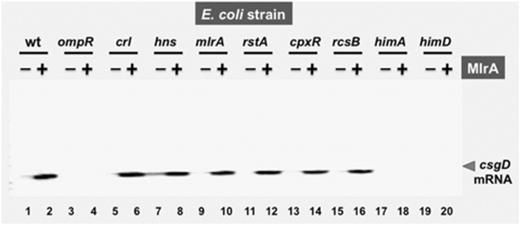
Effect of MlrA on the csgD promoter: primer extension analysis. Escherichia coli wild-type BW25113 and the csgD mutant carrying either pBADmlrA (lanes 2, 4, 6, 8, 10, 12, 14, 16, 18 and 20) or the vector (pBAD18) (lanes 1, 3, 5, 7, 9, 11, 13, 15, 17 and 19) were grown in YESCA medium in the presence of 0.02%l-arabinose. In mid-exponential phase growth at OD600 nm=0.4–0.5, total RNAs were prepared as described, and subjected to primer extension assay.
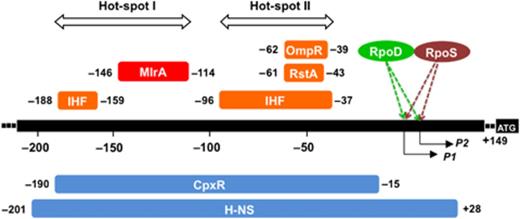
Location of the transcription factor-binding sites along the csgD promoter. The binding sites of MlrA, OmpR, RstA, CpxR, IHF and H-NS were determined by DNase-1 footprinting assays (Fig. 2; and Ogasawara et al., 2010). Five factors (OmpR, RstA, IHF, CpxR and H-NS) bind to the promoter-proximal hot-spot II, while four factors (IHF, MlrA, CpxR and H-NS) bind to the promoter-distal hot-spot I. P1 and P2 represent the transcription initiation site P1 (Hammar et al., 1995) and P2 promoter (Ogasawara et al., 2007a), respectively. Numbers shown at the ends of each regulator-binding site represent the distance from the P1 initiation site.
Promoter P1 is known to be recognized by RpoS sigma factor (Ogasawara et al., 2007a), and the activity of RpoS sigma is controlled by an accessory protein Crl (Bougdour et al., 2004). The induction level of csgD mRNA by MlrA overexpression in both the rpoS (data not shown) and the crl mutant (Fig. 4, lane 6) was as high as that in wild-type E. coli (Fig. 4, lane 2). This is not unexpected given that the csgD promoter P1 is recognized by both RpoD and RpoS (Ogasawara et al., 2007a). Escherichia coli contains an uncharacterized protein YcgE that exhibits an overall similarity of 76% to MlrA (Brown et al., 2001). We also tested possible functional replacement of MlrA by YcgE, but overexpression of YcgE did not affect the csgD mRNA level (data not shown). Together, these observations indicate that MlrA is a DNA-binding positive regulator of csgD transcription, but its homologue YcgE is not involved in csgD regulation.
Both the reporter assay and primer extension analysis indicated the stimulatory role of MlrA on csgD transcription. If this is the case, the set of genes under the control of the CsgD global regulator must be activated in the presence of MlrA overexpression. This possibility was tested by investigating the curli production level by staining of E. coli colonies with CR (Hammar et al., 1995). CR staining of E. coli colonies was not observed for the mlrA mutant (data not shown), supporting the prediction that curli fimbriae were not produced in the mlrA mutant.
Interplay between MlrA, OmpR and IHF
Three positive factors, IHF, OmpR and RstA, can associate simultaneously within the promoter-proximal hot-spot II region of transcription factor binding (Fig. 6), and cooperate with each other for activation of the csgD promoter. On the other hand, two negative factors, CpxR and H-NS, also bind to the same region and collaborate with each other (Ogasawara et al., 2010). As the enhancement of csgD mRNA synthesis by overproduction of MlrA was not observed in the ompR and ihf mutants, we then examined the possible interplay between MlrA, OmpR and IHF. The results indicated that MlrA binds in the spacer region between promoter-distal transcription factor-binding hot-spot I (including IHF-site I) and promoter-proximal hot-spot II (including IHF-site 2), to which OmpR also binds (Fig. 6). Gel shift assays using the CD6 probe DNA indicated that each of MlrA, OmpR and IHF alone formed CD6 complexes (Fig. 5a and b, lanes 2–11). In pair-wise assays, MlrA was found to bind together with either OmpR or IHF (Fig. 5a and b, lanes 12–16). In the simultaneous presence of MlrA, OmpR and IHF, all three regulators bind to the same CD6 probe forming MlrA–OmpR–IHF–DNA quaternary complexes (Fig. 5c). Together, we concluded that the three positive regulators, MlrA, IHF and OmpR, function independently, and do not show strong cooperation.
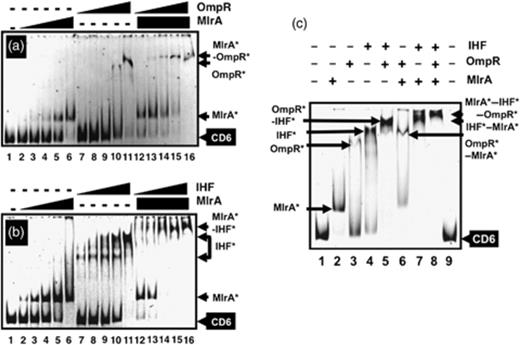
Simultaneous binding of three positive regulators, IHF, OmpR and MlrA. (a) Fluorescently labelled CD6 probe was incubated at 37°C for 30 min with MlrA (lanes 2–6: 0.625, 1.25, 2.5, 5.0 and 10 pmol) or OmpR (lanes 7–12 and 13–16: 2.5, 5.0, 10, 15 and 20 pmol), and directly subjected to PAGE. OmpR-binding assay was performed as above but in the presence of a fixed amount of MlrA (10 pmol). (b) Fluorescently labelled CD6 probe was incubated at 37°C for 30 min with MlrA (lanes 2–6: 0.625, 1.25, 2.5, 5.0 and 10 pmol) or IHF (lanes 2–12 and 13–16: 0.16, 0.31, 0.63, 1.25 and 2.5 pmol), and directly subjected to PAGE. IHF-binding assay was performed as above but in the presence of a fixed amount of MlrA (10 pmol). (c) Fluorescently labelled CD6 probe was incubated at 37°C for 10 min with IHF (5.0 pmol), MlrA (10 pmol) or OmpR (20 pmol) as indicated (+) above the gel panel, and then directly subjected to PAGE.
Discussion
Plasmid-encoded regulatory protein MerR was isolated as a mercury ion resistance gene (Ni' Bhriain et al., 1983; Lund et al., 1986; Heltzel et al., 1987). The MerR family of prokaryotic transcriptional activators have been identified in various bacteria except for E. coli and have a common molecular design, but have evolved to recognize and respond to different metals (Barkay et al., 2003; Brown et al., 2003; Hobman, 2007). MerR controls transcription of a set of genes (the mer operon) conferring mercury resistance. Homodimeric MerR represses transcription in the absence of mercury and activates transcription upon Hg(II) binding (Guo et al., 2010). One unique property of MerR is its ability to underwind DNA, resulting in activation of the target promoters by modulating the distance between promoter −35 and −10 (O'Halloran et al., 1989; Ansari et al., 1992). In addition, MerR was suggested to make interact directly with RNA polymerase (Kulkarni & Summers, 1999; Brown et al., 2003) as in the case of other class-I and class-II transcription factors (Igarashi & Ishihama, 1991; Ishihama, 1992, 1993; Busby & Ebright, 1999). MlrA contains a conserved N-terminal DNA-binding domain present in members of the MerR family, implying the mode of MlrA action should be the same with that of other MerR family transcription factors.
Here we identified the binding site of MlrA within the promoter-distal transcription factor-binding hot-spot I region of the csgD promoter (Fig. 6). MlrA binds a 33-bp-long palindromic sequence (see Fig. 3), which is much longer than the hitherto identified binding sequences by E. coli transcription factors (Ishihama, 2010) and probably induces DNA underwinding as in the case of MerR. DNA curvature induced by MlrA should influence not only the activity of neighbouring DNA such as binding affinity to positive factors, IHF, OmpR and RstA, and/or to negative fractors, H-NS and CpxR, but also the mode of the molecular interplay between csgD promoter-bound transcription factors.
The metal response regulator MerR interacts and binds heavy metals using its C-terminal domain. The C-terminal domain MlrA, however, exhibits little similarity to the MerR family regulators, indicating that MlrA interacts with an as yet unidentified effector using the C-terminal domain. Identification of the putative effector affecting MlrA activity is also important for detailed understanding of the regulatory role of MlrA in activation of the csgD promoter.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Priority Area System Cell Engineering by Multi-Scale Manipulation (17076016) to A.I., Grants-in-Aid for Scientific Research (A) (21241047) and (B) (18310133) to A.I., and Grant-in-Aid for JSPS Fellows (218850) to H.O. from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We also acknowledge the support from Project of Micro-Nano Technology Research Center of Hosei University. H.O. is a recipient of a JSPS Post-doctoral Fellowship.
References
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1.Escherichia coli strains used.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Author notes
Editor:David Clarke

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}